Cmax Pharmacology: Master It Now! (Side Effects?)
Plasma concentration, a key factor in dose optimization, heavily influences the understanding of cmax pharmacology. This is because Cmax, representing the maximum drug concentration observed post-administration, significantly impacts therapeutic efficacy and the potential for adverse events. Understanding the principles of cmax pharmacology is crucial not only for researchers at institutions like the FDA, which regulates drug safety and efficacy, but also for clinicians who prescribe medication. The area under the curve (AUC), another important metric, reflects total drug exposure but Cmax provides critical information about peak concentrations and subsequent effects. Mastering cmax pharmacology requires a solid grasp of these principles in order to mitigate side effects and maximize therapeutic benefit.
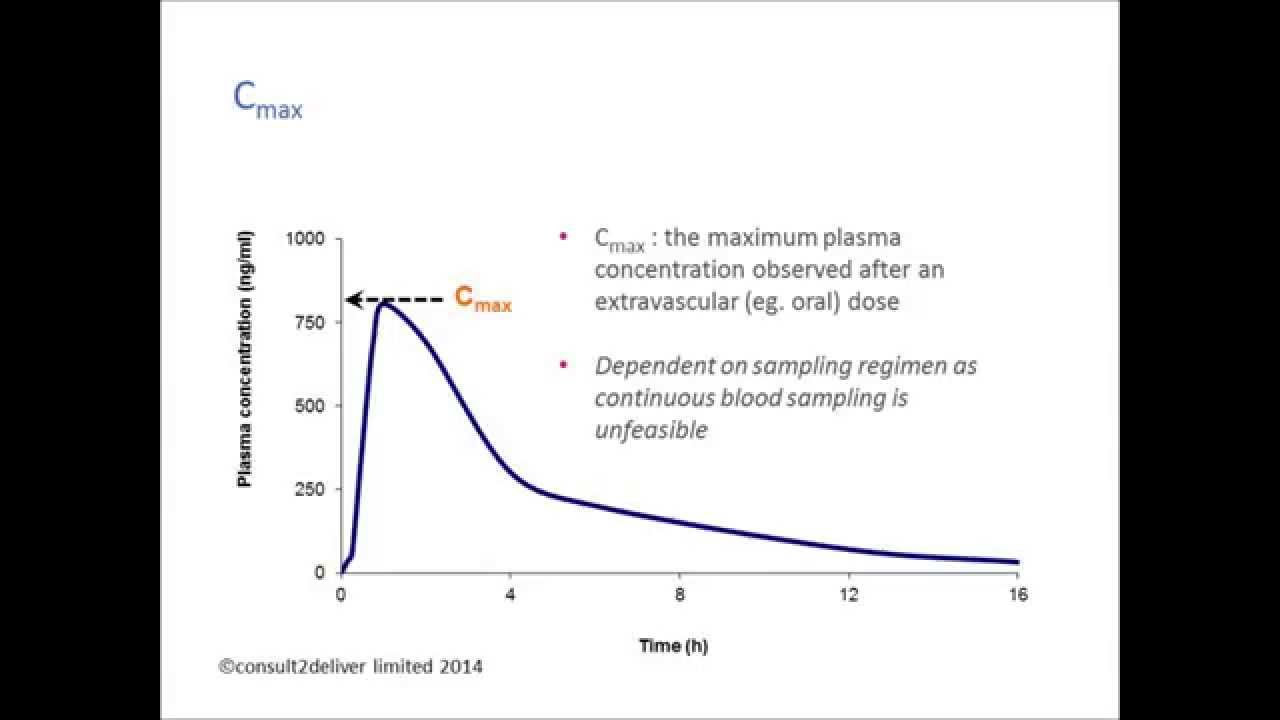
Image taken from the YouTube channel consult2deliver , from the video titled Pharmacokinetics: Why is Cmax important? .
Pharmacology, at its core, is the study of drugs and their effects on living systems. It encompasses a vast landscape, from understanding how drugs interact with cellular targets to elucidating their impact on entire organisms. The ultimate purpose of pharmacology is to develop and utilize medications safely and effectively to treat and prevent disease.
Within this expansive field, a critical parameter known as Cmax, or maximum concentration, holds particular significance. Cmax represents the highest concentration of a drug achieved in a specified compartment (usually plasma/serum) after the drug has been administered.
Defining Cmax: A Cornerstone of Drug Action
Cmax is not merely a data point; it is a key indicator of a drug's behavior within the body. It provides crucial information about the rate and extent of drug absorption, offering insights into how quickly and completely a drug enters the bloodstream. This, in turn, dictates the drug's ability to reach its target site and exert its therapeutic effect.
Understanding Cmax is paramount because it directly impacts both therapeutic efficacy and patient safety. Too low a Cmax might render a drug ineffective, while an excessively high Cmax can lead to toxicity.
Cmax and Pharmacokinetics: A Dynamic Relationship
Cmax is inextricably linked to pharmacokinetics (PK), the study of how the body processes a drug. PK encompasses the processes of absorption, distribution, metabolism, and excretion (ADME), all of which collectively determine the concentration-time profile of a drug in the body.
Cmax is a direct result of these ADME processes working in concert. Absorption determines how much and how quickly the drug enters the systemic circulation. Distribution governs the drug's movement to various tissues. Metabolism and excretion dictate the removal of the drug from the body.
By analyzing Cmax in conjunction with other PK parameters, such as the time to reach Cmax (Tmax) and the area under the concentration-time curve (AUC), we gain a comprehensive understanding of a drug's journey through the body.
Roadmap to Understanding Cmax
This section serves as a foundation for a deeper exploration of Cmax. We will delve into the determinants of Cmax, examining how factors like drug formulation, route of administration, and individual patient characteristics influence this crucial parameter. We will also explore the therapeutic relevance of Cmax, highlighting its importance in achieving desired clinical outcomes and minimizing the risk of adverse effects. Finally, we will address the potential for side effects associated with excessively high Cmax values, emphasizing the importance of careful dose optimization and patient monitoring.
The Pharmacokinetic Principles Underlying Cmax
Having established the fundamental significance of Cmax, it's crucial to understand the intricate processes that govern its value. The body's interaction with a drug is a dynamic interplay of absorption, distribution, metabolism, and excretion (ADME), collectively known as pharmacokinetics. These processes directly dictate the concentration-time profile of a drug, with Cmax representing a critical point on that curve. Let's dissect these pharmacokinetic principles to understand how they shape Cmax.
Drug Absorption: The Gateway to Systemic Circulation
Absorption is the process by which a drug moves from its site of administration into the systemic circulation. The rate and extent of absorption are primary determinants of Cmax. A faster absorption rate generally leads to a higher Cmax, while a greater extent of absorption results in a higher overall drug exposure.
Several factors influence drug absorption:
Route of Administration
The route of administration profoundly impacts absorption. Intravenous (IV) administration bypasses absorption entirely, resulting in immediate and complete bioavailability (F=1) and often the highest Cmax achievable.
Oral administration, conversely, involves a complex journey through the gastrointestinal tract, where absorption can be affected by gastric emptying, intestinal motility, and first-pass metabolism.
Other routes, such as intramuscular (IM), subcutaneous (SC), and transdermal, offer varying absorption rates and extents depending on the drug's properties and the specific formulation.
Drug Formulation
The formulation of a drug—whether it's a tablet, capsule, solution, or suspension—plays a crucial role in its dissolution and subsequent absorption. Tablets and capsules must disintegrate and dissolve before the drug can be absorbed.
Solutions, on the other hand, are already in a dissolved state and are generally absorbed more rapidly. Modified-release formulations, such as extended-release tablets, are designed to control the rate of drug release and absorption, often resulting in a lower Cmax compared to immediate-release formulations.
Physiological Factors
Physiological factors, such as gastric pH, intestinal motility, and the presence of food, can also influence drug absorption. Gastric pH can affect the ionization and solubility of drugs, impacting their ability to cross biological membranes.
Intestinal motility can influence the time available for absorption, while the presence of food can either enhance or inhibit absorption depending on the drug and its interactions with food components.
Drug Distribution: Reaching the Target Site
Following absorption, a drug is distributed throughout the body, moving from the bloodstream to various tissues and organs. Drug distribution plays a crucial role in determining drug concentrations at the site of action, which in turn influences the drug's therapeutic effect and potential for toxicity. While Cmax specifically refers to the maximum concentration in a defined compartment (usually plasma), the distribution characteristics of a drug influence how rapidly and extensively it reaches other tissues, impacting the overall concentration-time profile.
Factors influencing distribution include:
Tissue Binding
The extent to which a drug binds to tissues affects its distribution. Drugs with high tissue binding tend to have lower plasma concentrations and may exhibit a lower Cmax in plasma, even though their overall exposure might be significant due to accumulation in tissues.
Blood Flow to Tissues
Tissues with higher blood flow receive a greater amount of drug more quickly. This can lead to higher drug concentrations in well-perfused organs, potentially affecting both therapeutic and toxic effects.
Physiological Barriers
Physiological barriers, such as the blood-brain barrier (BBB), can limit the distribution of certain drugs to specific compartments. Drugs that cannot readily cross the BBB will have lower concentrations in the brain, impacting their ability to treat central nervous system disorders.
Volume of Distribution (Vd)
Volume of distribution (Vd) is a pharmacokinetic parameter that reflects the extent to which a drug distributes throughout the body. A large Vd indicates that the drug is extensively distributed into tissues, while a small Vd suggests that the drug remains primarily in the bloodstream. Vd is inversely related to Cmax: a drug with a large Vd will generally have a lower Cmax because it is dispersed throughout a larger volume.
Drug Metabolism: Altering the Drug's Structure
Drug metabolism, also known as biotransformation, is the process by which the body chemically modifies drugs. This process can affect drug concentrations over time, influencing both the rate of rise and fall of drug concentrations. Metabolism can lead to the formation of metabolites that are either more or less active than the parent drug.
Metabolism contributes to reducing Cmax by decreasing the amount of parent drug available in the systemic circulation. First-pass metabolism, which occurs in the liver and gut wall before a drug reaches systemic circulation after oral administration, can significantly reduce bioavailability and Cmax.
Enzyme systems involved in drug metabolism:
Cytochrome P450 Enzymes (CYPs)
Cytochrome P450 (CYP) enzymes are a family of enzymes that play a major role in drug metabolism. Different CYP enzymes are responsible for metabolizing different drugs, and variations in CYP enzyme activity can lead to significant differences in drug response.
Genetic Variability
Genetic variability in enzyme activity can result in individuals being classified as poor, intermediate, extensive, or ultra-rapid metabolizers. This can greatly affect drug concentrations, therapeutic efficacy, and the risk of adverse effects. For example, poor metabolizers may experience higher Cmax values and an increased risk of toxicity, while ultra-rapid metabolizers may have lower Cmax values and reduced therapeutic efficacy.
Drug Excretion: Eliminating the Drug from the Body
Drug excretion is the process by which the body removes drugs and their metabolites. Excretion affects both the duration and height of Cmax. Efficient excretion leads to a faster decline in drug concentrations and a shorter duration of action.
Renal and Hepatic Clearance Mechanisms
Renal clearance involves the removal of drugs and metabolites from the body via the kidneys. Glomerular filtration, tubular secretion, and tubular reabsorption are the primary renal excretion mechanisms. Hepatic clearance involves the removal of drugs and metabolites from the body via the liver. Biliary excretion is the primary hepatic excretion mechanism.
Clearance (CL)
Clearance (CL) is a pharmacokinetic parameter that reflects the efficiency of drug removal from the body. Clearance is inversely related to Cmax and AUC: a drug with a high clearance will generally have a lower Cmax and a shorter duration of action.
Bioavailability: The Fraction Reaching Systemic Circulation
Bioavailability (F) is the fraction of an administered dose of a drug that reaches the systemic circulation unchanged. It is a crucial pharmacokinetic parameter that directly influences the achievable Cmax value.
A drug with high bioavailability will have a higher Cmax compared to a drug with low bioavailability, assuming the same dose and rate of administration. Factors affecting bioavailability, such as first-pass metabolism and incomplete absorption, can significantly impact Cmax.
In conclusion, Cmax is a complex pharmacokinetic parameter that is influenced by a multitude of factors related to drug absorption, distribution, metabolism, excretion, and bioavailability. A thorough understanding of these pharmacokinetic principles is essential for optimizing drug therapy and minimizing the risk of adverse effects.
Having explored the individual pharmacokinetic processes that shape the drug concentration profile, we now shift our focus to the practical application of this knowledge. Understanding how Cmax is determined, interpreted, and predicted is essential for optimizing drug therapy and ensuring patient safety.
Calculating, Interpreting, and Predicting Cmax
Determining Cmax from Concentration-Time Curves
Cmax is directly observable from a concentration-time curve, which plots the drug concentration in plasma or serum against time after administration. It is visually identified as the highest point on this curve.
Creating a concentration-time curve involves collecting serial blood samples at various time points after drug administration and then measuring the drug concentration in each sample using analytical techniques such as liquid chromatography-mass spectrometry (LC-MS).
The time at which Cmax occurs (Tmax) is also recorded.
The shape of the concentration-time curve is influenced by the interplay of absorption, distribution, metabolism, and excretion. A steep ascending phase indicates rapid absorption, while a gradual descending phase reflects elimination processes.
The Relationship Between AUC and Cmax
While Cmax represents the peak concentration, the Area Under the Curve (AUC) reflects the total drug exposure over a given period. AUC is calculated by integrating the concentration-time curve.
AUC is dependent on the dose administered and the drug's clearance rate. A higher AUC indicates greater overall exposure to the drug.
Cmax and AUC, though distinct, are related. A higher Cmax doesn't necessarily mean a higher AUC, and vice versa. A drug with rapid absorption and elimination might have a high Cmax but a smaller AUC compared to a drug with slower absorption and elimination.
The ratio of Cmax to AUC can provide insights into the rate of drug absorption and elimination.
Influence of Half-Life (t1/2) on Cmax and Duration
The half-life (t1/2) of a drug is the time it takes for its concentration in plasma to decrease by half. Half-life is primarily determined by the drug's clearance and volume of distribution.
A drug with a short half-life will be eliminated quickly, leading to a shorter duration of action and potentially a lower Cmax if absorption is not rapid.
Conversely, a drug with a long half-life will persist in the body for a longer time, potentially resulting in a prolonged duration of action and a sustained, though not necessarily higher, Cmax.
The half-life influences not only the duration of drug action but also the frequency of dosing required to maintain therapeutic concentrations.
Understanding the Concentration-Time Curve
The concentration-time curve is a graphical representation of the drug's journey through the body, reflecting the dynamic interplay of pharmacokinetic processes.
It provides a visual summary of drug absorption, distribution, metabolism, and excretion.
Analyzing the shape of the curve, including the Cmax, Tmax, and AUC, allows for a comprehensive understanding of the drug's pharmacokinetic profile and its expected clinical effects.
The curve can also reveal potential issues, such as delayed absorption or rapid elimination, which may necessitate dosage adjustments.
Understanding concentration-time curves is critical for both drug development and clinical practice.
Having explored the individual pharmacokinetic processes that shape the drug concentration profile, we now shift our focus to the practical application of this knowledge. Understanding how Cmax is determined, interpreted, and predicted is essential for optimizing drug therapy and ensuring patient safety.
The Therapeutic Window: Linking Cmax to Clinical Outcomes
The ultimate goal of drug therapy is to achieve the desired therapeutic effect without causing unacceptable adverse reactions. This delicate balance is often described as the therapeutic window, a range of drug concentrations within which the probability of desired clinical response is high and the probability of unacceptable toxicity is relatively low. Cmax plays a crucial role in determining whether a drug concentration falls within this therapeutic window.
Cmax and the Therapeutic Range
The therapeutic range is the concentration of a drug in the blood that is associated with a high likelihood of therapeutic effect and a low probability of adverse effects.
This range is drug-specific and is determined through preclinical and clinical studies.
Cmax is a key determinant of whether a drug concentration reaches the lower end of the therapeutic range, thereby initiating a therapeutic effect. If the Cmax is too low, the drug concentration may never reach the minimum effective concentration (MEC), resulting in therapeutic failure.
Conversely, if the Cmax exceeds the upper limit of the therapeutic range, the risk of adverse effects increases significantly. Therefore, achieving a Cmax within the therapeutic range is essential for optimizing drug therapy.
Dosage Regimen and Cmax
The dosage regimen, encompassing the dose, frequency, and route of administration, is the primary determinant of Cmax. Adjusting these parameters allows clinicians to fine-tune drug concentrations and maintain them within the therapeutic window.
-
Dose: Increasing the dose generally leads to a higher Cmax. However, it's crucial to consider the potential for non-linear pharmacokinetics, where a disproportionate increase in Cmax may occur with increasing doses.
-
Frequency: More frequent dosing can lead to higher average drug concentrations and potentially higher Cmax values. However, careful consideration must be given to the drug's half-life (t1/2) to avoid accumulation and toxicity.
-
Route of Administration: The route of administration significantly impacts the rate and extent of drug absorption, directly influencing Cmax.
Intravenous (IV) administration typically results in the highest and most rapid Cmax, while oral administration is often associated with a lower and delayed Cmax.
The choice of route should be guided by the drug's properties, the desired speed of onset, and the patient's condition.
Cmax: Onset, Intensity, and Duration of Drug Effects
Cmax is intrinsically linked to the onset, intensity, and duration of a drug's effects. A higher Cmax generally leads to a faster onset and a greater intensity of effect, but potentially also a shorter duration, depending on the drug's elimination rate.
The onset of action is the time it takes for the drug concentration to reach the MEC. A higher Cmax achieved more quickly will result in a faster onset of action.
The intensity of effect is related to the peak drug concentration achieved at Cmax. Within the therapeutic range, a higher Cmax may correlate with a stronger therapeutic effect.
The duration of action is the length of time that the drug concentration remains above the MEC. While Cmax influences the initial attainment of therapeutic concentrations, the drug's elimination rate primarily determines how long the drug concentration remains within the therapeutic range.
Therefore, optimizing Cmax is crucial for achieving the desired balance between therapeutic efficacy and safety. Careful consideration of the pharmacokinetic principles governing Cmax, coupled with appropriate dosage regimen adjustments, is essential for maximizing the benefits of drug therapy and minimizing the risk of adverse effects.
Having established how Cmax relates to achieving therapeutic goals, it's equally important to acknowledge its potential downsides. While a sufficient Cmax ensures drug efficacy, exceeding optimal levels can dramatically increase the risk of adverse events, turning a therapeutic agent into a potential toxin. Understanding this relationship is critical for safe and effective drug use.
Cmax and the Risk of Adverse Effects
The relationship between Cmax and adverse effects represents a critical aspect of drug safety. While achieving a sufficient Cmax is essential for therapeutic efficacy, excessively high Cmax values can significantly increase the risk of adverse events, sometimes with serious consequences. This section explores this 'dark side' of Cmax, highlighting examples, monitoring strategies, and the role of drug interactions in exacerbating toxicity.
Elevated Cmax and Increased Adverse Effect Risk
A high Cmax doesn't automatically equate to toxicity, but it substantially raises the probability. Many drugs exhibit a concentration-dependent toxicity, meaning the higher the concentration, the greater the likelihood and severity of side effects.
This relationship can arise from several mechanisms:
- Receptor Saturation: At high concentrations, the drug may saturate its intended receptors and begin interacting with unintended targets, leading to off-target effects.
- Enzyme Inhibition: Some drugs, at high concentrations, can inhibit enzymes involved in their own metabolism or the metabolism of other drugs, leading to a build-up and prolonged exposure.
- Direct Cellular Toxicity: Certain drugs can directly damage cells at high concentrations, particularly in sensitive organs like the liver or kidneys.
Understanding the specific mechanisms by which a high Cmax leads to adverse effects is crucial for developing strategies to mitigate these risks.
Examples of Cmax-Related Adverse Reactions
Several drugs vividly illustrate the link between elevated Cmax and specific adverse reactions:
- Aminoglycoside Antibiotics (e.g., Gentamicin): These antibiotics are effective against severe bacterial infections. However, high Cmax levels are associated with ototoxicity (hearing damage) and nephrotoxicity (kidney damage). Monitoring Cmax is crucial to prevent these irreversible adverse effects.
- Tricyclic Antidepressants (TCAs): While used to treat depression, TCAs have a narrow therapeutic window. Elevated Cmax can lead to cardiac arrhythmias, seizures, and anticholinergic effects (e.g., dry mouth, blurred vision, urinary retention).
- Opioid Analgesics (e.g., Morphine): Effective for pain relief, opioids can cause respiratory depression at high Cmax levels, potentially leading to fatal outcomes. Co-administration with other CNS depressants further increases this risk.
- Digoxin: Used in the treatment of heart failure and certain arrhythmias, Digoxin toxicity can manifest as nausea, vomiting, visual disturbances (yellow halos), and potentially life-threatening arrhythmias.
These examples underscore the need for careful dosage titration and, in some cases, therapeutic drug monitoring to maintain Cmax within a safe range.
The Importance of Cmax Monitoring
Monitoring Cmax is essential for minimizing toxicology risks, especially for drugs with:
- A narrow therapeutic window.
- Known concentration-dependent toxicities.
- Significant inter-patient variability in pharmacokinetics.
Therapeutic drug monitoring (TDM) involves measuring drug concentrations in plasma at specific times after administration. This allows clinicians to:
- Ensure that Cmax falls within the therapeutic range.
- Adjust the dosage regimen based on individual patient characteristics.
- Identify potential drug interactions that could alter Cmax.
TDM is particularly valuable in vulnerable populations, such as:
- Pediatric patients: Whose organ function is still developing.
- Elderly patients: Who may have reduced organ function and multiple comorbidities.
- Patients with renal or hepatic impairment: Who may have altered drug clearance.
Regular monitoring of Cmax and other relevant clinical parameters empowers clinicians to make informed decisions and prevent serious adverse events.
Drug Interactions and Their Impact on Cmax and Toxicity
Drug interactions can significantly alter Cmax and, consequently, the risk of toxicity. These interactions can occur through various mechanisms:
- Absorption Interactions: Some drugs can inhibit or enhance the absorption of other drugs, altering the amount of drug that reaches systemic circulation and affects Cmax.
- Metabolism Interactions: Many drugs are metabolized by cytochrome P450 (CYP) enzymes. Co-administration of drugs that inhibit or induce CYP enzymes can dramatically increase or decrease the Cmax of other drugs, respectively.
- Distribution Interactions: Competition for plasma protein binding sites can lead to increased free drug concentrations and a higher Cmax.
- Excretion Interactions: Some drugs can inhibit renal transporters involved in drug excretion, leading to increased Cmax and prolonged exposure.
Clinicians must be aware of potential drug interactions and consider their impact on Cmax and toxicity. Reviewing a patient's medication list, including over-the-counter drugs and supplements, is crucial for identifying potential interactions and implementing appropriate monitoring or dosage adjustments. Being proactive and knowledgeable about drug interactions can significantly improve patient safety and prevent adverse outcomes.
Having established how Cmax relates to achieving therapeutic goals, it's equally important to acknowledge its potential downsides. While a sufficient Cmax ensures drug efficacy, exceeding optimal levels can dramatically increase the risk of adverse events, turning a therapeutic agent into a potential toxin. Understanding this relationship is critical for safe and effective drug use.
Regulatory Oversight: Cmax in Drug Development and Approval
The journey of a drug from the laboratory to the pharmacy shelf is a long and complex one, heavily governed by regulatory agencies. Among the many pharmacokinetic parameters scrutinized, Cmax holds a prominent position, acting as a key indicator of both drug efficacy and safety. This section examines how regulatory bodies, particularly the Food and Drug Administration (FDA) in the United States, utilize Cmax data in the drug approval process.
FDA's Role in Setting Cmax Guidelines
The FDA plays a critical role in ensuring that all drugs marketed in the US are both safe and effective. As part of this responsibility, the agency establishes guidelines and requirements related to Cmax during drug development and approval.
These guidelines are not necessarily rigid limits but rather benchmarks and considerations that inform the FDA's assessment of a drug's risk-benefit profile. The FDA doesn't dictate a specific Cmax value for a drug; instead, it evaluates Cmax within the context of the drug's overall pharmacokinetic and pharmacodynamic profile, target population, and intended use.
The evaluation considers:
- Efficacy: Does the achieved Cmax consistently correlate with the desired therapeutic effect in clinical trials?
- Safety: Is the Cmax associated with an acceptable level of adverse events? Are there subpopulations particularly vulnerable at certain Cmax levels?
- Dose Proportionality: Does the Cmax increase proportionally with increasing doses, or is there a non-linear relationship suggesting potential for saturation or toxicity?
The FDA uses these considerations to inform labeling requirements, dosage recommendations, and post-market surveillance strategies. It often requires manufacturers to provide extensive data on Cmax, including population pharmacokinetic (PopPK) modeling, to understand how Cmax varies across different patient groups (e.g., based on age, weight, sex, renal function, etc.).
Clinical Trials: Unveiling Safe and Effective Cmax Levels
Clinical trials are the cornerstone of drug development, providing the crucial data needed to establish safe and effective Cmax levels. These trials, conducted in phases, systematically evaluate the drug's pharmacokinetic properties, efficacy, and safety in human subjects.
Phase I trials often focus on assessing the drug's safety and pharmacokinetic profile, including Cmax, in a small group of healthy volunteers.
Phase II trials explore the drug's efficacy and dose-response relationship in a larger group of patients with the target condition. Here, Cmax is carefully monitored to identify the optimal dose range that balances efficacy and tolerability.
Phase III trials involve large, randomized, controlled studies designed to confirm the drug's efficacy and monitor adverse effects in a real-world setting. These trials provide a comprehensive assessment of Cmax variability and its relationship to clinical outcomes across diverse patient populations.
- Dose-Response Studies: These studies are crucial for determining the relationship between dose, Cmax, and therapeutic effect.
- Drug-Drug Interaction Studies: Assessing the impact of co-administered drugs on Cmax is essential for identifying potential safety concerns.
- Special Population Studies: Evaluating Cmax in vulnerable populations, such as the elderly or those with renal or hepatic impairment, is critical for tailoring dosage recommendations.
Ultimately, the data gleaned from clinical trials informs the FDA's decision-making process, allowing the agency to establish appropriate labeling and dosage guidelines that ensure patients receive the maximum benefit from a drug while minimizing the risk of harm. The rigorous scrutiny of Cmax, from initial development through post-market surveillance, underscores its pivotal role in safeguarding public health.
Video: Cmax Pharmacology: Master It Now! (Side Effects?)

FAQs: Understanding Cmax Pharmacology
This section answers common questions about Cmax pharmacology, helping you better understand its significance and implications.
What exactly does Cmax represent?
Cmax stands for maximum drug concentration. In cmax pharmacology, it's the highest concentration of a drug observed in the blood, serum, or plasma after administration. It's a crucial indicator of drug absorption and bioavailability.
Why is Cmax important in drug development?
Cmax is important because it directly correlates with the likelihood of a drug achieving its therapeutic effect. Understanding cmax pharmacology also helps predict potential side effects, as high concentrations can sometimes lead to toxicity.
How does the timing of Cmax affect treatment?
The time it takes to reach Cmax (Tmax) is also crucial. A shorter Tmax means the drug reaches its peak concentration faster, potentially leading to a quicker onset of action. This is especially relevant for pain medications or drugs requiring rapid relief.
Can Cmax values be affected by individual patient factors?
Yes, cmax values are indeed influenced by factors like age, weight, kidney and liver function, and even genetics. These patient-specific variables can alter drug absorption, distribution, metabolism, and excretion, ultimately affecting the observed Cmax in cmax pharmacology.